General aspects of modeling and computer simulations
Contents
General aspects of modeling and computer simulations¶
-Last updated on Thursday, March 24 2022 at 14:35
Learning goals:
To have an overview of the process involved in modeling and simulation. This is illustrated by a concrete example. Please notice that although the example is taken from a particular field, the process itself doesn’t depend on the application field.
To understand how modeling even simple systems can be a very challenging and interdisciplinary problem.
Keywords: physical phenomena, algorithm, code, model, analysis, debugging, comparison to experiments, visualization
Big Science. Hallelujah. Big Science. Yodellayheehoo.
You know. I think we should put some mountains here.
Otherwise, what are all the characters going to fall off of?
And what about stairs? Yodellayheehoo. Ooo coo coo.—Laurie Anderson, Big Science
Music: Laurie Anderson, Big Science
General approach¶
Let’s discuss a few general and important aspects that relate to all computer simulations and modeling. Fig. 20 shows the general idea - this is independent of the application field.

Fig. 20 Some general aspects about modeling and computer simulations. Building a model and setting up simulations is a complex process and each stage needs to be considered carefully and validated extensively. Mistake at any level can render the whole process wrong with heaps of meaningless data. That happens more frequently that one would expect; that the software doesn’t crash doesn’t imply anything about the correctness of the results. The figure is based on [2]¶
Example: Modeling water¶
Let’s consider the items in Fig. 20 using a concrete example. The procedure and considerations below are, by necessity, simplified but should give an idea the type of considerations one has to take. Some of the choices and decisions are also coupled.
Physical phenomenon/phenomena: Let’s use the behavior of bulk water as an example. We need to decide what we wish to model. Liquid-gas or liquid-ice phase transition? Structure of water? Or perhaps diffusion of water or the dynamics of the hydrogen bond network? Or hydrophobic hydration? One also needs to review previous research to know what kind of problems there have been and what are the undisputed correct results (if any).
Example: Water dynamics is complex & has practical importance:
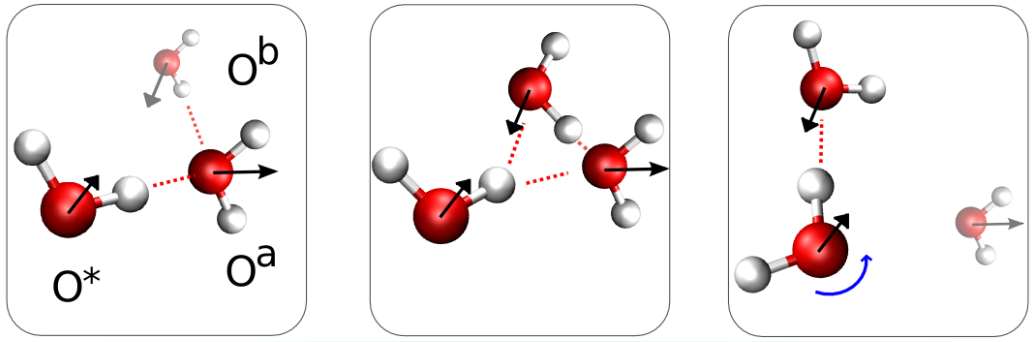
Some interesting previous results:

Construct/choose a model: We need to decide what is the level, that is, the level of detail, and time and length scales that we are interested in. Do we need quantum mechanics? Is a classical atomistic model enough or should we use a coarse-grained model? Even at the classical level there are tens of existing models including acronyms such as ST2, TIP3P, TIP4P, TIP4P/2005, TIP5P, TIP5P/2008, SPC, SPC/E and so on. How do we know which one(s) to choose? Should one use more than one model? What has been done with these models? Is it/are they able to capture the phenomena of interest?
Here: A quantum level model was needed. The basic model didn’t include van der Waals interactions (density functional theory doesn’t) so they needed to be added. This may sound odd, but inclusion of van der Waals interactions in a quantum level simulation is a non-trivial task.

Choose numerical algorithm(s): Even in the case we are planning to use a software package, we need to decide which one of the algorihms the package offers we should use. For example, for integration of the equations of motions each package offers several options. If we are going to write our own code, then we need to decide which one to implement. How do we deal with interactions? They need to be truncated but what is the most reliable way to do it? Does the system have long-range interactions? If so, do we wish to use Fourier transform-based algorithms or real space ones? In either case, there are several choises that one needs to understand. If we are writing out own code, we need to decide how to speed up the simulations - normal MD scales as \(\mathcal{O}(N^2)\) where \(N\) is the number of particles (the so-called big-O notation is used to express how well algorithms scale). We can do better using tables or multisteping. Which one to choose? Do we need constant temperature or pressure constant pressure to better mimic experimental conditions? Which method to choose? What kind of boundary conditions do we need? How can we simulate bulk conditions with a relatively small number of particles? If one uses a method that is part of an existing code, then the choice of the algorithm comes with the software.

Write/choose a simulation program: Do we need to write our own code or can we use one the existing packages? This problem is very general and we could choose a software package. There are lots of possibilities including CPMD, CP2K, Gromacs, NAMD, Amber, CHARMM, DL Poly, LAMMPS and others. Or should we write your own code? If so, which programming language to use? Fortran, C, C++, python, CUDA or something else? Will this program be used in other projects? How much time will it take to write a code, debug and verify it? Do we wish to provide as open source? If so, how will it be maintained? Debugging is the most time consuming and important part!
In this case, we are interested in the picosecond-level dynamics and wish to use a quantum mechanical model. Of the many possible choices, let’s choose the CPMD package (Car-Parrinello Molecular Dynamics). It also turned out that the code needed some additions. They were tested and verified against previous simulations and experiments.

Perform computer simulations: Before starting simulations: Have you debugged your code or/and checked that the package that you have chosen is appropriate and has no errors in the parts relevant for you? Do you understand the input data and parameters, and the simulation protocol? Important: even the most well-established codes have errors and problems, but the biggest error source of all is the user! Finally, how to run very long runs in batches?
Here: We can prepare the systems on local workstations, but the production simulations need large-scale resources with lot of memory. Compute Canada comes to help.

Analyze data and interpret: This is where the interesting part starts - provided we have confidence in our simulations. Do we need to write software to analyze the data or do appropriate tools exist? If the former, there are also practical questions such as how to read in the data; the data files may be massive and they may use some specific format(s), or they may be compressed.
Here, we can analyze hydrogen bonding in bulk water and in the solvation shell of a small hydrophobic group in a molecule. It is not all four-coordinated. Some of the analysis software needed to be written.
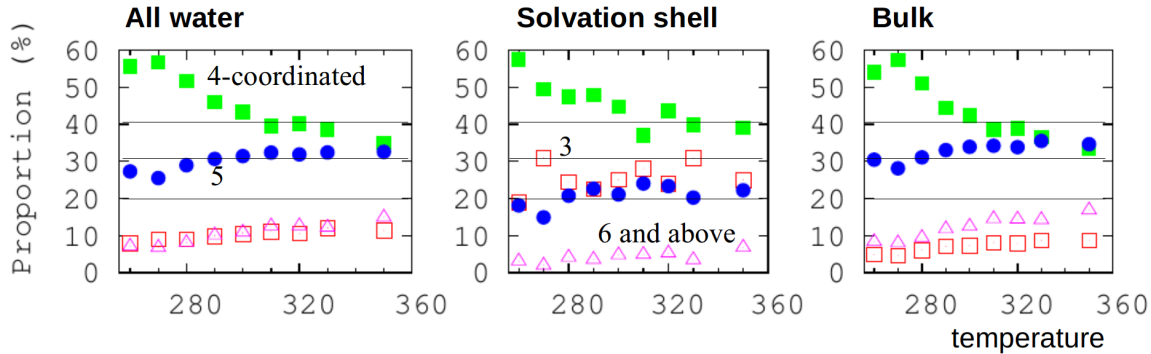
Hydrophobicity: effect of density and order on water’s rotational slowing down [10]

And one should always create visualizations:
The video below shows a close-up from an ab initio simulation of water. Hydrogen bonds that form and break are shown as dashed lines. See also how the vibrations of the bonds change when the bonding changes. This is a closeup from a simulation of 54 water molecules using Born-Oppenheimer Molecular Dynamics (BOMD). Visualization using VMD (Visual Molecular Dynamics) [4], simulation using the CPMD (Car-Parrinello Molecular Dynamics) software.
Additional questions: What kind of resources are available? How long will the simulations take, that is, how large systems and for how long time can be simulated. Is that enough? How about data storage abd backup, is there enough? Do we need long term data storage?
Summary¶
In the above, there are several terms and concepts that are new. We will encounter them when we set up and perform simulations. The aim here has been to give a rough idea of the process of modeling works: Simply downloading a well-known software package and using the default parameters and inputs is not acceptable. The software will run, but there is no guarantee that the results will be correct if one doesn’t understand the inputs and outputs. In addtion, the results will not be relevant (even if correct) if one doesn’t have a good research question. Reproducing data is not research. The software is not to blame for any of those matters, it does what the user tells it do. The user is the source of most errors; even if/when there is a bug/are bugs (and there are always are some), it is the user’s responsibility to test and ensure the validity of results. If bugs are found in a published software, one should notify the developers with precise information including test results.
Additional information for those interested: Water models¶
To put the example above to further perspective and to give an idea how complex water the simple looking humble water molecule is, below are lists of water models at different levels of description. None of the lists are by no means exhaustive and its purpose is simply to give an idea of the complexity of the problem - textbooks tend to give a simplified view. Water, as simple as it is, remains to be very difficult to model correctly. Some recent reviews are provided for example by [1, 3, 6, 7, 8]
Historical:¶
W.C. Röntgen - the same person who discovered x-rays (and was the recipient of the 1901 Nobel Prize in Physics), developed a two species model (1897). The water molecules were classified into fluid-like and ice-like. J.D. Bernal and R.H. Fowler developed a model with tetrahedral geometry (1933). This is the model that forms the basis of current understanding. Linux Pauling’s (Nobel Prize in Chemistry 1954 and Nobel Prize in Peace 1962)) model established clathrate structure (1935) and the model of John Pople (Nobel Prize in Chemistry 1998) showed hydrogen bond bending (1951).
Quantum mechanical:¶
All the approaches have their own models. Importantly, however, van der Waals interactions are not part of the standard quantum mechanical approach and they have to be added separately. As a side note the van der Waals interactions are named after J.D. van der Waals, the recipient of the 1910 Nobel Prize in Physics.
Classical MD level:¶
There are two main families of water models:
TIP = Transferrable Intermolecular Potential
SPC = Simple Point Charge
To given an idea of the abundance of models, here are some without explanations: BF, ST2, TIPS, TIPS2, TIP3P, TIP3Pm, TIP3P/Fs TIP4P, TIP4P-Ew, TIP4P/2005, TIP4P/2005f, TIP4P/\(\varepsilon\), TIP4P/FQ, TIP4P-HB, TIP4P-i, TIPTP/ice, TIP4P-pol, TIP4PQ, TIP4P-QDP, TIP4P-D, TIP5P, TIP5P-E, TIP5P/2019, OPC, SPC, SPC/E, SPC/\(\varepsilon\), SPC/A, SPC/F, SPC/F2, SPC/FQ, SPC/Fw, SPC-pol, SPC/HW.
We will use some of these models during this course.
Coarse-grained:¶
There are also models that descrive water in a larger scale. The idea is that one so-called coarse-grained molecule somehow approximates a bundle of 3 or 4 actual water molecules. Here are some: MARTINI, MARTINI polarized, ELBA, SIRAH, DPD, Mercedes-Benz, BMW, mW.
References¶
- 1
Emiliano Brini, Christopher J. Fennell, Marivi Fernandez-Serra, Barbara Hribar-Lee, Miha Lukšič, and Ken A. Dill. How water's properties are encoded in its molecular structure and energies. Chemical Reviews, 117(19):12385–12414, sep 2017. doi:10.1021/acs.chemrev.7b00259.
- 2
R. W. Eastwood and J. W. Hockney. Computer Simulation Using Particles. Adam Hilger, 1988.
- 3
Aziz Ghoufi and Patrice Malfreyt. Calculation of the surface tension of water: 40 years of molecular simulations. Molecular Simulation, pages 1–9, aug 2018. doi:10.1080/08927022.2018.1513648.
- 4
William Humphrey, Andrew Dalke, and Klaus Schulten. VMD: visual molecular dynamics. Journal of Molecular Graphics, 14(1):33–38, Feb 1996. doi:10.1016/0263-7855(96)00018-5.
- 5
Damien Laage and James T. Hynes. A molecular jump mechanism of water reorientation. Science, 311(5762):832–835, Feb 2006. doi:10.1126/science.1122154.
- 6
Alexey V. Onufriev and Saeed Izadi. Water models for biomolecular simulations. Wiley Interdisciplinary Reviews: Computational Molecular Science, pages e1347, nov 2017. doi:10.1002/wcms.1347.
- 7
Jeremy C. Palmer, Peter H. Poole, Francesco Sciortino, and Pablo G. Debenedetti. Advances in computational studies of the liquid-liquid transition in water and water-like models. Chemical Reviews, 118(18):9129–9151, aug 2018. doi:10.1021/acs.chemrev.8b00228.
- 8
Lars Gunnar Moody Pettersson, Richard Humfry Henchman, and Anders Nilsson. Water—the most anomalous liquid. Chemical Reviews, 116(13):7459–7462, jul 2016. doi:10.1021/acs.chemrev.6b00363.
- 9
Y. L. A. Rezus and H. J. Bakker. Observation of immobilized water molecules around hydrophobic groups. Physical Review Letters, Oct 2007. doi:10.1103/PhysRevLett.99.148301.
- 10
John Tatini Titantah and Mikko Karttunen. Hydrophobicity: effect of density and order on water's rotational slowing down. Soft Matter, 11(40):7977–7985, 2015. doi:10.1039/c5sm00930h.